Microbiology Spectrum | 检测转录因子结合位点的新方法
中科院深圳先进院合成所助理研究员倪磊、研究员金帆为共同通讯作者。

文章上线截图
文章链接:https://doi.org/10.1128/spectrum.03783-22
转录因子能够以特定序列与基因专一性结合,保证目的基因以特定的强度在特定的时间空间表达。转录因子和DNA的相互作用在基因调控网络(GRNs)中起着核心作用。目前检测转录因子-DNA结合特异性的主要方法需要先对转录因子结合的DNA片段进行捕获富集然后再进行测序(或其他分析),如染色质免疫沉淀 (ChIP)、配体系统进化指数富集(SELEX)和DNA亲和纯化测序(DAP-Seq)等。这些方法涉及复杂的实验程序,如基因组DNA的碎片化和扩增、转录因子的免疫沉淀等,较高的操作门槛不利于新手或跨学科人员快速展开实验。
在本研究中,金帆团队开发了一种名为AIDmut-Seq的体内方法。活化诱导胞苷脱氨酶(AID)可以将单链DNA序列中的碱基C脱氨化形成碱基U, DNA复制后发生C-T或G-A替代。研究人员将AID融合到转录因子上,并在体内诱导融合蛋白表达,这样就可以在转录因子结合位点附近引入突变,后续可以通过全基因组测序直接检测。由于不需要对转录因子结合的片段进行捕获富集,而仅需对突变标记进行测序,因此AIDmut-Seq的整个工作流程仅包含细菌培养、基因组提取和生信分析三个步骤(图 1),不涉及其他复杂的实验操作,大大节省了实验人员的时间和劳动成本。
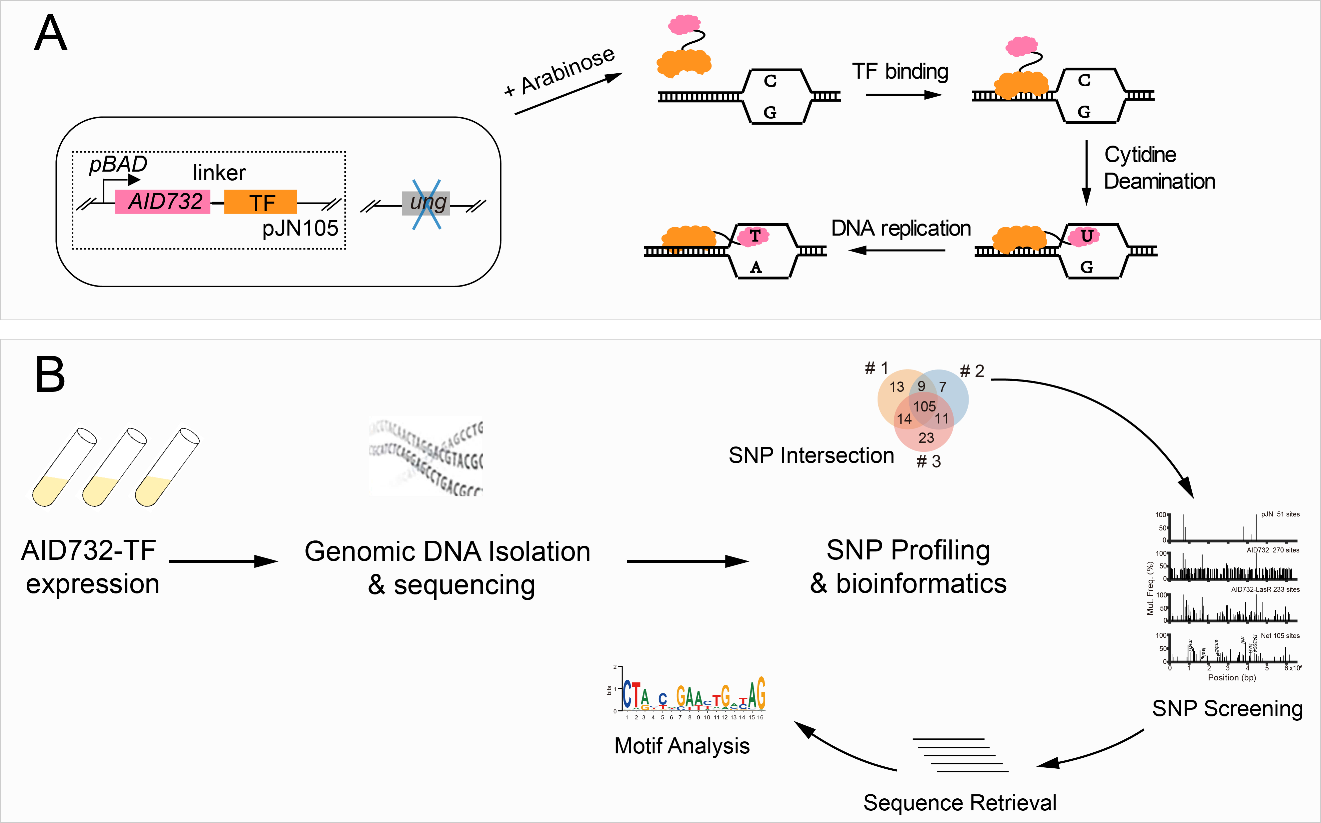
图1 AIDmut-Seq的原理和步骤
研究人员使用不同类型的转录调节因子对AIDmut-Seq进行了验证,表明该方法对大多数转录激活因子(如LasR,FleQ,ErdR,GacA,ExsA等)的具有较高效率。而对一些小的转录抑制因子(如RsaL和AmrZ等)虽然表现出较低的效率,但通过该方法计算得到的转录因子识别基序(motif)和现有方法得到的基序具有很大相似性。此外,使用AIDmut-Seq还得到了许多之前没有发现的转录因子结合位点和新的调节模式。这些结果证明了AIDmut-Seq的高效性和泛用性,可以作为现有方法的重要补充工具(图2)。

图2 AIDmut-Seq适用于大多数转录因子
研究人员将ADImut-Seq与目前最常用的ChIP-seq进行了“标杆测试”(benchmarking)。比较发现, AIDmut-Seq和ChIP-seq对TFBS具有相似的检测精度,但AIDmut-seq具有更窄的检测窗口。尤其对于启动子上存在多个结合位点的位置,AIDmut-Seq相比ChIP-seq具有更好的分辨率,因此AIDmut-Seq在识别同一启动子中的多个结合位点具有潜在的优势(图3)。
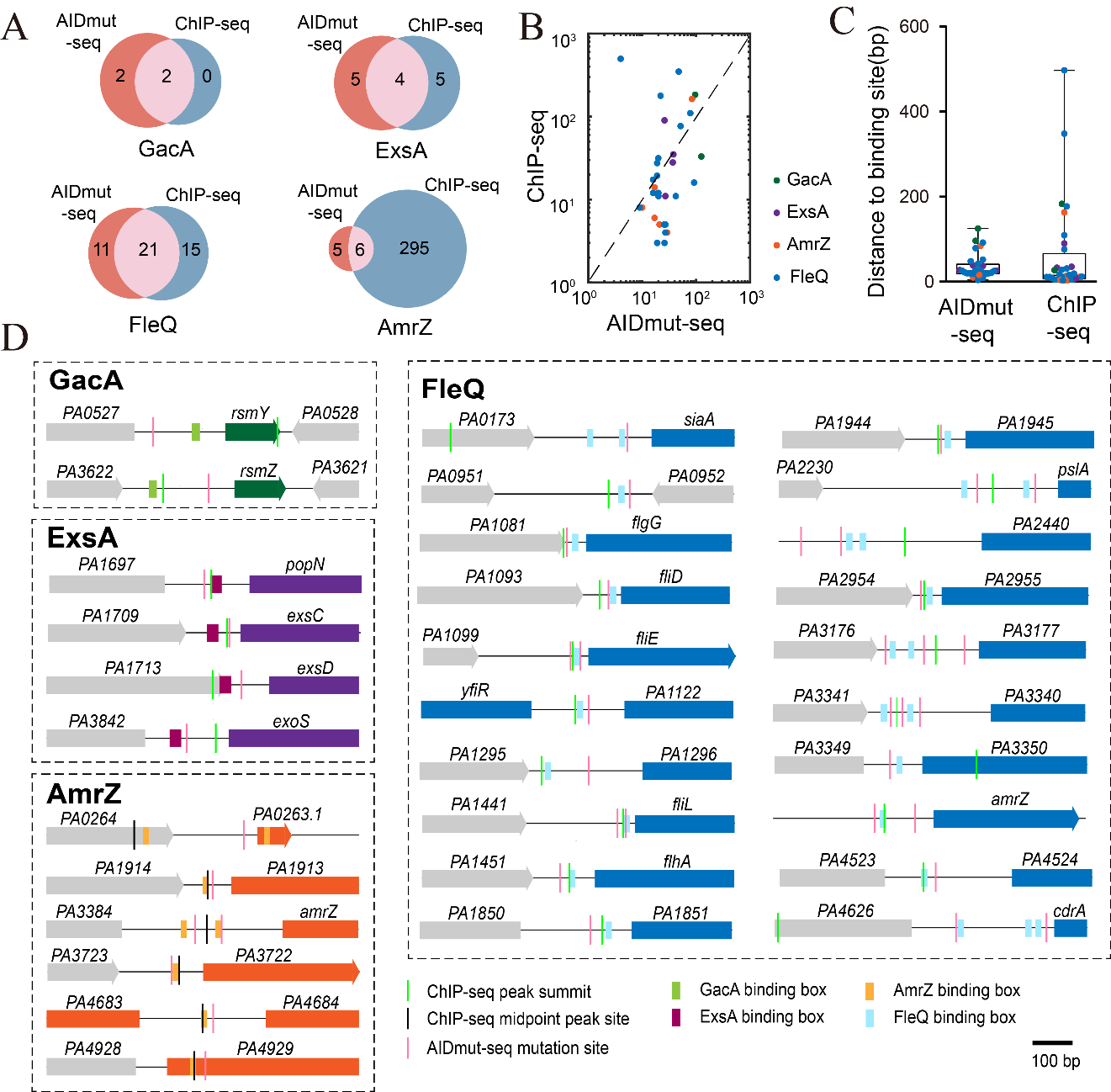
图3 AIDmut-Seq与ChIP-seq的比较
该研究得到了科技部重大研发计划、国家自然科学基金、中国科学院科学仪器开发和深圳合成生物学创新研究院等项目支持。